隨著工業(yè)的發(fā)展����,汞污染越來(lái)越嚴(yán)重。由于汞的污染特性�����,不易分解及轉(zhuǎn)移����。造成了食品中富集了各種含汞化合物,從而導(dǎo)致各種慢性�����、急性汞中毒事件頻出����。目前,食品中汞的主要存在形式可分為無(wú)機(jī)汞和有機(jī)汞兩大類����,其中有機(jī)汞的毒性遠(yuǎn)大于無(wú)機(jī)汞,無(wú)機(jī)汞主要是對(duì)腎臟有毒性作用����,而有機(jī)汞主要對(duì)腦組織傷害比較大。因此對(duì)汞形態(tài)的監(jiān)測(cè)顯得尤為重要。
本文參考國(guó)家衛(wèi)生和計(jì)劃生育委員會(huì)發(fā)布的《GB 5009.17-2014 食品中甲基汞的測(cè)定》�����,通過HPLC-ICPMS聯(lián)用��,采用C18柱��,利用等度洗脫的方式對(duì)稀酸浸提后的大米進(jìn)行了分析�,同時(shí)對(duì)樣品采取了加標(biāo)處理,且加標(biāo)回收率良好�����。測(cè)試結(jié)果表明����,該方法能夠快速準(zhǔn)確地進(jìn)行大米樣品中汞形態(tài)的分析。
1.儀器簡(jiǎn)介
本方法采用Skyray Instrument LC-310E與ICP-MS 2000E聯(lián)用����,采用一鍵式數(shù)據(jù)采集、精準(zhǔn)式譜圖定位及強(qiáng)大的軟件譜圖處理功能���,可滿足不同的形態(tài)測(cè)試需求(如圖1)�。

圖1.HPLC-ICPMS聯(lián)用測(cè)汞形態(tài)的分析儀
2.測(cè)試原理
樣品溶液經(jīng)過前處理,由HPLC進(jìn)樣口進(jìn)樣�,經(jīng)色譜柱分離��,通過霧化器霧化后送入高溫等離子體中����,干燥、原子化����、電離,元素離子經(jīng)接口室進(jìn)入質(zhì)譜儀����,通過離子透鏡系統(tǒng)、質(zhì)量分析器及檢測(cè)器�,檢測(cè)器對(duì)相應(yīng)元素離子做出響應(yīng)(每秒離子計(jì)數(shù)cps(counts
per second),經(jīng)軟件處理����,響應(yīng)強(qiáng)度和時(shí)間組成的峰面積與相應(yīng)組分離子濃度成正比關(guān)系進(jìn)行定性定量分析。
3.實(shí)驗(yàn)部分
3.1實(shí)驗(yàn)設(shè)備及試劑
液相色譜-電感耦合等離子體質(zhì)譜聯(lián)用儀(江蘇天瑞儀器股份有限公司)���;
電子天平(BSA224S�,賽多利斯);
色譜柱(Ultimate XB-C18�,4.6×150mm,5μm)��;
超純水系統(tǒng)(Millipore�,電阻率為18.25MΩ·cm);
鹽酸(優(yōu)級(jí)純��,Scharlau)���;
氨水(優(yōu)級(jí)純����,上海晶純?cè)噭┯邢薰荆?
乙酸銨(優(yōu)級(jí)純���,阿拉丁化學(xué)試劑)����;
L-半胱氨酸(生物純���,阿拉丁化學(xué)試劑)��;
甲醇(色譜純�,Oceanpak 色譜純?cè)噭?
汞形態(tài)標(biāo)準(zhǔn)溶液(水中汞GBW(E)083186、甲基汞GBW(E)083364�、乙基汞GBW(E)081524,中國(guó)計(jì)量科學(xué)院)�;
超聲波(KH-500E,昆山禾創(chuàng));
雙功能水浴恒溫振蕩器(SHA-B,江蘇國(guó)華)����;
高速離心機(jī)(TG16G,凱特)�。
3.2樣品前處理
稱取大米樣品1.0g(精確至0.001g),置于15mL的離心管中��,加入10mL的鹽酸溶液(5mol/L)靜置過夜�����。后經(jīng)常溫水浴振蕩60min����,于4℃下以8000r/min的轉(zhuǎn)速進(jìn)行離心15min。取上層清液2mL至5mL的容量瓶中�,用稀氨水調(diào)節(jié)PH到7.0,再加入0.1mL的L-半胱氨酸(10g/L)����,最后用超純水定容至刻度并搖勻���。用0.45μm的有機(jī)系濾膜過濾后,待測(cè)�����。
3.3流動(dòng)相及汞形態(tài)標(biāo)準(zhǔn)溶液配制
3.3.1流動(dòng)相的配制
60mmol/L乙酸銨+0.1%L-半胱氨酸+5%甲醇:稱取4.627g乙酸銨�,1.21gL-半胱氨酸于100mL容量瓶中,加入少量的超純水并于電熱板上加熱使其完全溶解��,冷卻后定容至100mL���。將定容好的溶液移入1000mL的淋洗液瓶中��,同時(shí)加入50mL的甲醇����,定容至1L���。經(jīng)0.45μm的有機(jī)系濾膜過濾后���,于超聲波超聲脫氣30min,現(xiàn)用現(xiàn)配��。
3.3.2汞形態(tài)標(biāo)準(zhǔn)溶液:
三種汞標(biāo)準(zhǔn)溶液采用淋洗液按照梯度濃度的方式依次被稀釋為1.0μg/L、2.0.μg/L�����、4.0μg/L���、8.0μg/L���、10.0μg/L 混合標(biāo)準(zhǔn)溶液,同時(shí)配制5μg/L三種汞形態(tài)的單標(biāo)溶液���,用來(lái)判別每種汞形態(tài)的出峰時(shí)間。
3.4實(shí)驗(yàn)條件
3.4.1色譜條件
色譜條件詳見表1�����。
表1. HPLC分析條件
|
參數(shù)
|
參數(shù)設(shè)定
|
|
色譜柱
|
Ultimate XB-C18�����,5μm�����,150×4.6mm。
|
|
流動(dòng)相
|
60mmol/L乙酸銨+0.1%L-半胱氨酸+5%甲醇(pH=7.0)���。
|
|
流 速
|
1.0mL/min
|
|
柱 溫
|
25℃
|
|
進(jìn)樣量
|
100μL
|
|
洗脫程序
|
等度洗脫
|
3.4.2質(zhì)譜條件
ICP-MS分析條件見表2�����。
表2.ICP-MS分析條件
|
參數(shù)
|
參數(shù)設(shè)定
|
參數(shù)
|
參數(shù)設(shè)定
|
|
功 率
|
1300W
|
等離子氣
|
13L/min
|
|
輔助氣
|
1.06L/min
|
載 氣
|
1.2L/min
|
|
采樣深度
|
16
|
分析模式
|
標(biāo)準(zhǔn)模式
|
|
待測(cè)質(zhì)量數(shù)
|
202
|
駐留時(shí)間
|
10ms
|
3.5實(shí)驗(yàn)結(jié)果
3.5.1標(biāo)準(zhǔn)曲線
按照混合標(biāo)準(zhǔn)溶液濃度1.0μg/L���、2.0.μg/L、4.0μg/L�����、8.0μg/L�����、10.0μg/L 進(jìn)樣得到色譜圖�����,計(jì)算其峰面積�����,以濃度和峰面積繪制標(biāo)準(zhǔn)曲線,線性相關(guān)系數(shù)均大于0.999���,結(jié)果詳見圖2-1~2-3��。
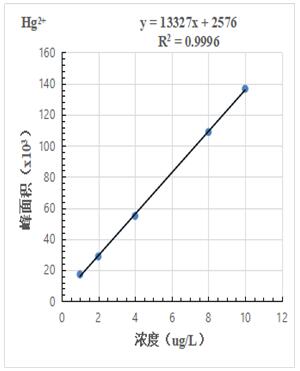
圖2-1.Hg2+標(biāo)準(zhǔn)曲線
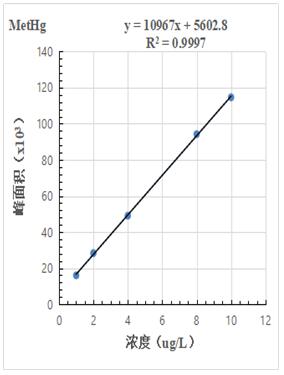
圖2-2.MetHg標(biāo)準(zhǔn)
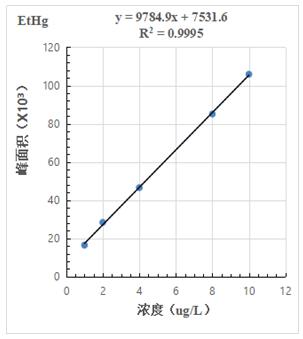
圖2-3.EtHg標(biāo)準(zhǔn)曲線
3.5.2色譜分離圖
1.0μg/L��、2.0.μg/L���、4.0μg/L、8.0μg/L����、10.0μg/L 汞形態(tài)混合標(biāo)準(zhǔn)溶液分離色圖譜如圖3所示,其中無(wú)機(jī)汞(Hg2+)��、甲基汞(MetHg)����、乙基汞(EtHg)相對(duì)保留時(shí)間分別為118s����、185s、379s,從譜圖疊加圖上可以看出���,三種汞形態(tài)出峰時(shí)間穩(wěn)定����,且分離度較好���。
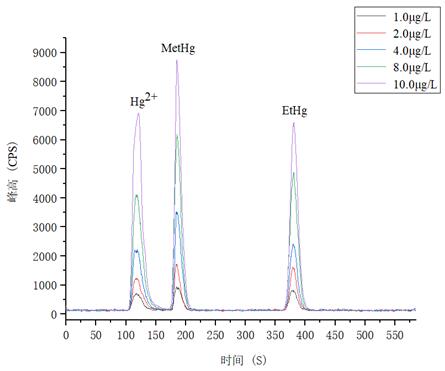
圖3. 汞形態(tài)混合標(biāo)準(zhǔn)溶液分離圖譜
3.5.3 檢出限
如圖4所示為1.0μg/L汞形態(tài)混和標(biāo)準(zhǔn)溶液分離圖譜�����,以各形態(tài)峰附近基線處的3倍信噪比(S/N)峰高對(duì)應(yīng)的濃度作為檢出限����,溶液中Hg2+�����、MetHg��、EtHg分別為0.112μg/L�、0.0718μg/L、0.0698μg/L���。
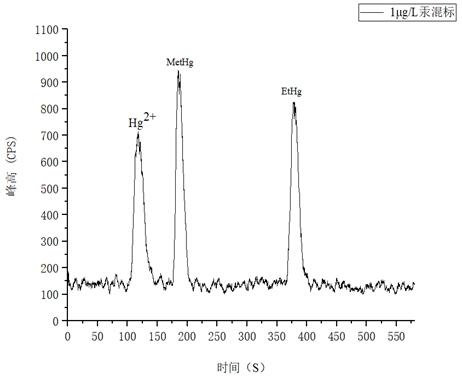
圖4. 1.0μg/L汞形態(tài)混合標(biāo)準(zhǔn)溶液圖譜
3.5.4 測(cè)試結(jié)果
以大米樣品為試樣�����,進(jìn)行稀酸浸提�����,同時(shí)為了考察方法的準(zhǔn)確性��,并對(duì)大米試樣采取了加標(biāo)的方式��,測(cè)試結(jié)果詳見表3��。
表3 大米樣品及樣品加標(biāo)結(jié)果
|
組分名稱
|
稀釋后濃度
|
原液濃度
|
加標(biāo)后濃度
|
加標(biāo)回收率
|
|
(μg/L)
|
(μg/kg)
|
(μg/L)
|
(%)
|
|
無(wú)機(jī)汞
|
0.227
|
5.68
|
5.84
|
112
|
|
甲基汞
|
未檢出
|
ND
|
4.50
|
90
|
|
乙基汞
|
未檢出
|
ND
|
4.67
|
93
|
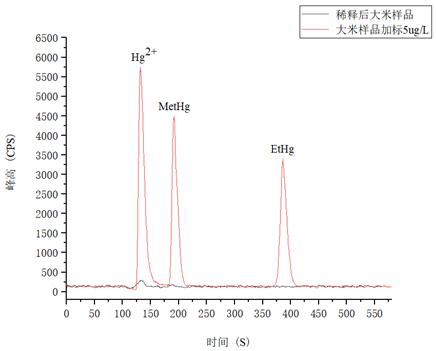
圖5. 大米樣品及樣品加標(biāo)5.0μg/L圖譜
4. 結(jié)論
本文建立了LC-ICPMS測(cè)試大米中汞形態(tài)的分析方法�����,結(jié)果表明該方法檢出限低����、測(cè)試結(jié)果準(zhǔn)確��,其加標(biāo)回收率在90%~112%之間����,線性相關(guān)系數(shù)均在0.999以上��;同時(shí)汞的三種形態(tài)能夠在8min左右完成�,且分離度較好�,該方法可滿足食品中大米樣品汞形態(tài)分析的要求。
5.參考文獻(xiàn)
[1] 國(guó)家衛(wèi)生和計(jì)劃生育委員會(huì)發(fā)布 GB 5009.17-2014食品安全國(guó)家標(biāo)準(zhǔn) 食品中甲基汞的測(cè)定 液相色譜-原子熒光光譜聯(lián)用方法���,2015�����。
